Geeniuuringutega viiruste jahil
-

Tartu Ülikooli meditsiinilise viroloogia kaasprofessor
-

Tartu Ülikooli meditsiinilise mikrobioloogia ja viroloogia kaasprofessor
-

Tartu Ülikooli meditsiinilise viroloogia teadur
-

Tartu Ülikooli bioinformaatika spetsialist
Kui kunagi peaks puhkema mõne uue tundmatu nakkuse epideemia, on meil praegusest olukorrast kaasa võtta piisavalt kogemusi.
Nakkushaigused on koos inimesega eksisteerinud alates selle liigi algusaegadest saadik. Siiski arvatakse, et laialdased epideemiad said alguse peamiselt ajal, kui inimesed hakkasid maad harima ja kariloomi pidama, ning inimeste asustustihedus tõusis üle kriitilise piiri. Modernse aja epideemiaidki on seostatud peamiselt inimpopulatsiooni kasvu, linnastumise ning kiirelt kasvanud rahvusvahelise reisimise ja kaubavahetusega.
Peamine viis, kuidas uued haigustekitajad inimesi nakatavad, on ülekanded loomadelt inimesele (zoonootiline levik). Sellise ülekande tõenäosust suurendab vahetu kokkupuutumine loomadega nagu küttimine, eksootiliste loomade ning nendest valmistatud toodete kaubandus ja elusloomade turud. Tõenäosus eri liiki haigustekitajate ülekandeks sõltub loomast, kust nakkus pärit, looma-inimese kontakti iseloomust ja sagedusest ning eelkõige patogeenist endast. Viimastel aastatel on selgunud, et patogeenide levimisel metsikust loodusest inimesele on oma roll ka kliimamuutustel. Selliste haigustekitajate nagu Dengue, Zika ja Lääne-Niiluse viiruse ülekandumist inimestele seostatakse just viirust ülekandvate sääskede ning puukide levikupiiride muutusega ning levimisega väljapoole endeemilisi alasid. Lisaks on võimalik senitundmatute patogeenide ilmumine aladelt, mis olid siiani püsinud inimtegevusest puutumatud (liustikud, džunglid jt).
Läbi ajaloo on epideemiate põhjustajateks olnud nii bakterid kui ka viirused. Bakteriaalsetest epideemiatest on tuntumad katku ja koolera puhangud. Katku põhjustajaks on Yersinia pestis ning tuntumad tema põhjustatud epideemiad olid Justinianuse katk ja must surm. Justinianuse katk sai alguse Egiptusest, levis Ida-Rooma impeeriumis ja selle naaberaladel 6. sajandil, surmates arvatavasti kuni 100 miljonit inimest. Must surm, mis levis 14. sajandil, sai alguse Ida-Aasiast ning levis läbi Kesk-Aasia ka Euroopasse. Selle tagajärjel hukkus 75–200 miljonit inimest. Teadaolevalt said esmased haiguse levikut takistavad meetmed alguse just musta surma ajal – kontrolliti linnadesse sisenemist, eraldati haiged tervetest, sulgeti sadamad nakatunud piirkondadest tulijatele ning pandi laevade meeskondi isolatsiooni.
Viimastel sajanditel on märkimisväärsemateks olnud Vibrio cholerae põhjustatud koolera epideemiad. Alates 19. sajandist on teada seitse suuremat epideemiat, mis on nõudnud umbes kolme miljoni inimese elu.
Viiruslikest epideemiatest on suurima mõjuga olnud gripi pandeemiad. Gripiviiruse A/H1N-i põhjustatud Hispaania gripp levis aastatel 1918–1920 ning viis hauda hinnanguliselt kuni 50 miljonit inimest. Pandeemia põhjustajaks arvatakse olevat linnugripi viirus, mis geneetilise adaptsiooni tagajärjel nakatas inimest. Läänemaailma suurimates linnades küll püüti gripi levikut piirata, propageerides inimestevahelise distantsi hoidmist, sulgedes koole, kirikuid ja teatreid ning keelates avalikke kogunemisi, kuid vastavate meetmetega jäeti Esimese maailmasõja tekitatud kaose tõttu lootusetult hiljaks. Praktiliselt võimatu oli takistada põgenike reisimist ja hoida piirikontrolli. Sõjavägede ümberpaiknemine ning sõdurite halvad elutingimused aitasid haiguse levikule ainult kaasa.
Viimastel kümnenditel on miljoneid inimelusid nõudnud ka HIV/AIDSi-epideemia. Tegemist on peamiselt vere- ja sugulisel teel leviva viirusega, mis sisenes inimpopulatsiooni juba 20. sajandi alguses, kuid saavutas laiema leviku alles 1960. aastatel. HIV peamine vorm on pärit šimpansidelt, kellel viirus olulist haigust ei tekita. Vaatamata efektiivsele HIVi vastasele ravile, mis võimaldab nakkust kandvatel inimestel elada suhteliselt normaalset elu, ei ole siiani suudetud välja töötada ei vaktsiini ega ravi, mis viiruse täielikult kehast eelmaldaks. Märkimisväärseid jõupingutusi selle pandeemia peatamiseks tehakse tänini.
KOROONAVIIRUSED INIMESTEL
Kuni sajandivahetuseni peeti inimeste koroonaviiruseid üsna süütuteks, peamiselt hooajalisi hingamisteede nakkusi põhjustavateks viirusteks. Selle ajani olid tuntud neist vähesed, tuntumad ehk 229E ja OC43, mille nakkuse peamisteks sümptomiteks on nohu, palavik, kurguvalu ja köha.
Paraku otsustas 2002. aasta näidata koroonaviiruste kurjemat palet. Aasias kirjeldati SARS-CoVi (Severe Acute Respiratory Syndrome Coronavirus). Viirus suutis nakatada üle 8000 inimese, kellest umbes kümme protsenti surid. Arvatakse, et viirus levis inimestele nahkhiirtelt, kuid tsiibetkassi vahendusel. Peale 2004. aastat pole uusi SARS-CoV juhte kirjeldatud, kuid arvestades tema reservuaari olemasolu looduses ei ole uued puhangud võimatud.
Veidi keerulisemaks osutus olukord MERS-CoViga (Middle East Respiratory Syndrome Coronavirus), mis avastati kümmekond aastat hiljem. Sarnaselt SARS-CoViga pärineb MERS-CoV samuti nahkhiirtelt, kuid tema vahendajateks ning ka reservuaariks võivad olla koduloomad kaamelid. Inimeselt inimesele levib MERS-CoV otsese kontakti kaudu ja tänu sellele on võimalik viiruse levikut karantiini abil kontrolli all hoida. Võrreldes SARS-CoViga on MERS-CoVi juhte teada vähem – praeguseks umbes 3000, kuid viirus on loomadelt inimesele kandunud korduvalt. Samuti on selle viiruse tõttu suremus oluliselt kõrgem – kuni kolmandik nakatunutest. Senised uurimused SARS-CoVi ja MERS-CoViga on tekitanud olulised teadmised patogeensetest koroonaviirustest üldisemalt. Välja on töötatud laboratoorsed metoodikad ning töövõtted nende viiruste uurimiseks, samuti on hangitud olulist infot nende viiruste vastaste vaktsiinide arendamiseks.
Nagu hästi teada sai SARS-CoV-2 epideemia alguse 2019. aasta lõpus Wuhani linnas Hiinas. 2021. aasta kevadeks oli pandeemia põhjustanud ülemaailmselt rohkem kui 150 miljonit nakkust ning üle kolme miljoni surma. Enamik Euroopa riike on SARS-CoV-2 epideemiast olnud väga tugevalt mõjutatud, olles diagnoositud nakatunute arvuga olnud mingil pandeemia hetkel tugevalt üle terve Euroopa keskmise. Ei Eesti ega kogu maailma meditsiinile ja majandusele pole seni ükski epideemia taolise suurusega väljakutseid varem esitanud.
VIIRUSE PALJUNEMINE JA MUTATSIOONIDE TEKE
Viirustel aitab ümbritseva keskkonnaga kohaneda muteerumine. Sarnaselt paljude teiste viirustega on ka SARS-CoV-2 paljunemine üsna robustne, mis tähendab, et viiruse paljunemise käigus ei vaevuta eriti põhjalikult kontrollima paljundatud geneetilise materjali vastavust algsele variandile. Mutatsioonid, mis SARS-CoV-2 paljunemisel tema genoomi tekivad, võib laias laastus jagada punktmutatsioonideks ning deletsioonideks. Enamik tekkivatest mutatsioonidest ongi punktmutatsioonid, mis tähendab, et vahetusse on läinud üks nukleotiid – näiteks mingis kindlas genoomi positsioonis olev A-nukleotiid on asendunud G-ga. Veidi harvem tekivad deletsioonid – need on mutatsioonid, kus teatud osa genoomist (tavaliselt küll hästi lühike) läheb kaduma. Võrreldes punktmutatsioonidega on selliste mutatsioonide mõju viiruse omadustele oluliselt suurem.
Funktsionaalselt võib mutatsioonid viiruse seisukohast jagada samuti eri gruppidesse: neutraalsed, kahjulikud ja kasulikud. Enamik mutatsioone on neutraalsed, mis tähendab, et viiruse omadused nende mutatsioonide ilmumisest ei muutu. Sellised mutatsioonid on tänuväärne materjal viiruste genoome jälgivatele teadlastele. Nende abil saab viiruse erinevaid tüvesid ja variante üksteisest eristada ja liigitada. Mingis mõttes moodustavad need mutatsioonid justkui viiruste sõrmejälje – see võimaldab kindlaks teha, kust vaadeldav viirus tuli ja kellega ta lähemalt suguluses on. Sageduselt järgmise hulga mutatsioonidest moodustavad viirusele kahjulikud mutatsioonid. Nendel pole mõtet pikemalt peatuda, kuna kahjulikke mutatsioone kandvad viirused surevad või jäävad halastamatus konkurentsis teistele viirustele kiirelt alla. Paraku suudavad viirused toota aga sedavõrd palju järglasi, et osade viiruste surm kahjulike mutatsioonide tõttu suuremat mõju ei avalda. Kõige harvemad on viirusele kasulikud mutatsioonid, kuid nende mutatsioonide võimalik mõju viirusele ja sellest johtuvalt ka tema peremehe käekäigule on kõige suurem.
Viirustel aitab ümbritseva keskkonnaga kohaneda muteerumine.
SARS-CoV-2 epideemia kontekstis muudavad viirusele kasulikud mutatsioonid kõige enam kahte viiruse omadust. Esiteks viiruse hulka patsiendis ning viiruse hulka, mis on vajalik uue peremehe nakatumiseks. Need mutatsioonid tekivad valdavalt viiruse ogavalgu geeni, mis vastutab viiruse seondumise eest tema retseptorile (ACE2) ning viiruse sisenemise eest peremehe rakku. Teiseks oluliseks omaduseks, mida SARS-CoV-2-l on kasulik kiirelt muuta, on nähtavus immuunsüsteemile. Inimese immuunsüsteem suudab tuvastada viirust peamiselt viiruse valkudes olevate lühikeste piirkondade põhjal. Kui nendesse piirkondadesse tekivad mutatsioonid, siis neid piirkondi ei pruugita enam ära tunda ja viirus muutub immuunsüsteemi jaoks nähtamatuks. Selliseid mutatsioone nimetatakse ka immuun-escape ehk immuunsüsteemi eest põgenemise mutatsioonideks. Need mutatsioonid võimaldavad viirusel põgeneda kahte olulisemat tüüpi viirusvastuse eest – antikehade vahendatud ning T-rakkude poolt vahendatud viirusvastuse eest. Siinkohal pole vahet, kas immuunvastus on tekkinud varasema nakkuse või vaktsineerimise käigus. Immuun-escape mutatsioonid on suures osas samuti koondunud viiruse ogavalku, kuna see valk on viirusosakese pinnal ja hästi kättesaadav antikehadele.
Hinnanguliselt on SARS-CoV-2 viiruse muteerumise kiirus üks-kaks mutatsiooni kuus ühe viiruse genoomi kohta. Kui võtta arvesse, et maailmas on 2021. aasta maikuu seisuga umbes 20 miljonit nakatunut ning SARS-CoV-2 genoomi pikkus on kõigest 30 000 nukleotiidi (võrdluseks inimese genoom on umbes 100 000 korda suurem), siis viirusel on suurepärane võimalus läbi proovida kõikvõimalikud mutatsioonid oma genoomis. See ongi viinud olukorrani, kus viirusel SARS-CoV-2 on välja kujunenud palju erinevaid tüvesid ja variante, millest mõned on nakkavamad kui teised ning millest mõned suudavad vähem või rohkem varjuda nii juba kord läbipõetud nakkuse kui ka vaktsineerimise tekitatud immuunvastuse eest.
OLULISEMAD SARS-CoV-2 VARIANDID JA TÜVED
SARS-CoV-2 puhul räägitakse enamasti variantidest, genotüüpidest või tüvedest. Ega väga head standardit nende mõistete kasutamiseks veel välja kujunenud pole. Osa virolooge ütlevad, et eri tüvedeks saab viiruseid kutsuda siis, kui nende tekitatud haiguspildid üksteisest erinevad. Vastasel juhul tuleb rääkida vaid variantidest või genotüüpidest. Paljud rahvusvahelised organisatsioonid, sealhulgas ECDC (European Center for Disease Prevention and Control) kasutavad ka liigitust VOI (variant of interest) ja VOC (variant of concern) – ehk siis huvi pakkuvad ja muret tekitavad variandid. VOCide ehk muret tekitavate variantide puhul on vähemalt mingitest uuringutest teada, et nad käituvad nakatumisel või haiguste tekitamisel kuidagi agressiivsemalt kui viiruse algne ehk nii-öelda metsikut tüüpi (wild type) Wuhani tüvi. VOI ehk huvipakkuva variandi puhul on esialgu õhus pigem kahtlus, et see viirus võiks agressiivsemalt käituda – näiteks tema genoomis on mingid mutatsioonid, mis on olemas ka mõnel VOC tüvel.
Epideemia ajal on SARS-CoV-2 viirustel ilmunud väga erinevaid mutatsioone ning tüvesid või variante. Tükk aega pärast epideemia algust levisid Euroopas vaid algsed, nii-öelda metsikut tüüpi Wuhani viirusele küllatki sarnased SARS-CoV-2 variandid. Euroopas suudeti pikalt ära hoida Aasias tekkinud uute mutantide ulatuslikumat sissetungi. Asjaolud muutusid, kui kohale jõudis viirus, mis kandis mutatsiooni D614G. See oli üks esimesi variante, mis suutis edukamalt levida kui „harilikud“ variandid. Selle mutatsiooniga viirused hõlmasid kiiresti kogu Euroopa ja levisid kiirelt ka ülejäänud maailmas. On teada, et nimetatud mutatsioon muudab viiruse ogavalgu struktuuri nii, et see seondub tõhusamalt rakkudega. See omakorda tähendab, et ilmselt on ka nakatumiseks vajalik doos madalam. Allpool nimetatud tüvedest ja variantidest kuuluvad kõik, VOCide ehk muret tekitavate viiruste hulka.
Vaieldamatult kurikuulsaim ja võib-olla ka kõige efektiivsem levija on SARS-CoV-2 tüvi nimega B.1.1.7 ehk nn Inglise tüvi (Inglismaal tuntud ka kui Kenti tüvi). Esimesena kirjeldati seda Inglismaal 20. septembril 2020 võetud proovist ning vaatamata piirangutele levis see kiiresti alguses Kenti ümbruses, sai peagi domineerivaks kogu Inglismaal ning hiljem suuremas osas Euroopas. Selle tüve Eesti-sisene levik tuvastati esmakordselt detsembri lõpul võetud proovides. Edasi läks nii nagu mujal maailmas – viiruse levikueelise tõttu kasvas tema osakaal pidevalt, kõige kiiremini vahemikus 2021. aasta veebruari algusest kuni aprilli alguseni (umbes 10% nädalas). Praeguseks on see saavutanud taseme üle 95 protsendi kõikidest juhtudest. Inglise tüve eristavad algsest Wuhani tüvest mitmed mutatsioonid, kuid olulisemaks peetakse neist kahte ogavalgu mutatsiooni. Deletsioon – del69/70 – on kahe aminohappe kadumine ogavalgu NTD (N-terminaalne domeen, deleteerib valgus aa 69 ja 70) piirkonnas. See suurendab nakatamisvõimet ilmselt tänu immuunsüsteemi eest pagemisele. Teine mutatsioon on N501Y, mis märgib ühe aminohappe asendumist teisega ogavalgu rakule seondumise piirkonnas. Selle mutatsiooni tulemusena seondub ogavalk jällegi tugevamalt ning seega vajab viirus nakatamiseks madalamat doosi. Mitmed uuringud on näidanud, et kuigi Inglise tüve levik on oluliselt efektiivsem ning tema haiguskulg võib olla raskem, siis vaktsineeritute kaitse kõigi Eestis kasutatavate vaktsiinide korral selle viiruse vastu peaks olema võrreldav „harilike“ tüvede omaga.
Esimene B.1.351 ehk Lõuna-Aafrika Vabariigi (LAV) tüve juhtum avastati 2020 oktoobri algul Lõuna-Aafrika Vabariigis. Aastavahetuse paiku raporteeriti selle tüve jõudmisest juba nii Euroopasse, Aasiasse, Austraaliasse kui ka Ameerikasse. Kuid enamikes riikides väljaspool Aafrikat ei ole sellel tüvel olnud levikueelist teiste tüvede ees ning tema osakaal on jäänud madalaks. Selle tüve olulisemateks mutatsioonideks on nagu Inglise tüvelgi N501Y ning lisaks E484K. See E484K mutatsioon vähendab „harilike“ tüvede vastu tekkinud antikehade seondumist. Eestis on LAV tüve korduvalt sisse toodud ning kirjeldatud on ka selle tüve kohapealset levikut. Siiski ei ole see suutnud siin massilisemalt levima hakata. Murelikuks teeb selle tüve puhul hoopis tema võimalik kõrvalehiilimine vaktsiinide tekitatud immuunkaitsest. Katseklaasi katsed on näidanud, et nn algsete SARS-CoV-2 tüvede vastu disainitud vaktsiinid ja nende esile kutsutud antikehad on mõnevõrra madalama efektiivsusega LAV tüve suhtes. Vaktsiinide toime päriselus LAV tüvede vastu pole päris selge, kuid peaks selguma õige pea. Värsked andmed on väga lootusrikkad, näidates, et vaktsiinide toime on efektiivne nii Inglise kui ka LAV tüve vastu.
Vaieldamatult kurikuulsaim ja võib-olla ka kõige efektiivsem levija on viiruse SARS-CoV-2 nn Inglise tüvi.
P1 ehk Brasiilia tüve kirjeldati esimest korda Brasiilias 2020. aasta novembri lõpus. Selle tüve ohtlikeks mutatsioonideks peetakse N501Y-d, E484K-d ja K417T-d. See tüvi tekitas ajakirjanduses ja teadlaskonnas väga palju elevust kui selgus, et ta suudab Manaus Brasiilias nakatada suurt hulka varem COVID-19 läbipõdenud elanikkonnast. Samas, selle juhtumi puhul on diskuteeritud palju ka eksitustest läbipõdenute protsendi hindamisel. Siiski on P1 tüve osakaal 2021. aasta mai alguses USAs tõusmas, kuid Euroopas esialgu veel minimaalne. Katseklaasikatsete põhjal on alust arvata, et see allub vaktsiinidele umbes sama hästi kui Inglise tüvi.
India tüvi (B.1.617.1) identifitseeriti esmalt 2020. aasta oktoobri algul Maharashtras Indias. Selle ohtlikeks mutatsioonideks peetakse L452R-i ja E484Q-d. Esimene neist, L452R, tagab parema seondumise rakule ning E484Q korral kahtlustatakse immuunsüsteemi eest pagemist nagu E484K-gi korral. Uuemad katseklaasi andmed viitavad siiski, et vaktsiinid töötavad selle tüve korral hästi ja et E484Q mõju antikehade aktiivsusele on oluliselt väiksem kui E484K korral. Kuigi India tüve arvukuse kasvuga langeb kokku aprillikuiste nakatumiste kasv Indias, siis tõenäoliselt pole põhjuseks mitte see konkreetne tüvi, vaid ilmselt ikkagi kevadised suured rahvahulkade kogunemised ja sealt alguse saanud nakkusahelad.
Ravimitööstused on juba katsetamas oma vaktsiinidele kordusdoose mitmete variantide ja tüvede vastu.
Kokkuvõtteks võib öelda, et uusi SARS-CoV-2 tüvesid ning neid tekitavaid mutatsioone tekib juurde kogu aeg. Osa neist tüvedest saavutavad ka suurema arvukuse, kuid kas selle põhjuseks on lihtsalt juhus, kiirem levimisvõime või oskuslikum immuunsüsteemi eest pagemine, on sageli väga keeruline öelda. Samas võib juba tekkinud ning laialdaselt levinud tüvedesse juurde tekkida uusi mutatsioone, mis panevad aluse omakorda uutele tüvedele. Kindlasti tuleb meeles pidada, et mida suurem on nakatunute arv kogu maailmas, seda kõrgem on tõenäosus, et mõni ebameeldiv tüvi tekib ja hakkab levima. Ilmselt joonistub erinevate tüvede ja erisuguste vaktsiinide omavaheline mõõduvõtt selgemalt välja alles siis, kui märkimisväärne osa populatsioonist on ära vaktsineeritud. Samas ravimitööstused on juba katsetamas oma vaktsiinidele kordusdoose erinevate variantide ja tüvede vastu.
VIIRUSE GENOTÜPISEERIMINE JA SEKVENEERIMINE
Viimase aastakümne tormiline areng genoomika ehk geneetilise materjali sekveneerimise (järjestamise) valdkonnas on tekitanud olukorra, kus organismide genoomide järjestusi saab määrata väga suures mahus. Alguse sai see üksikute bakterite ning inimese genoomi järjestuste kindlakstegemisest paarkümmend aastat tagasi. Praegu ollakse võimelised määrama suurte populatsioonide või isegi kõikide ühes keskkonnas olevate organismide DNA järjestusi (metagenoome). Erandiks pole siinkohal ka viirused. Kasutusele on võetud süvasekveneerimise meetodid, millega saab põhimõtteliselt kindlaks määrata iga üksiku DNA või RNA fragmendi järjestuse, mis uuritavas proovis on. Süvasekveneerimise puuduseks on aga sellega kaasnev tohutu andmemaht ja vajadus seda bioinformaatiliselt analüüsida. Ühest sekveneerimisreaktsioonist saadakse miljoneid või isegi miljardeid väikeseid genoomsete järjestuste fragmente, mida on vaja puhastada, süstematiseerida ja asetada nende õigetele kohtadele referentsjärjestustel.
MOLEKULAARNE EPIDEMIOLOOGIA ENNE SARS-CoV-2
Epidemioloogilisi uuringuid, kus kasutatakse geneetilist infot, nimetatakse molekulaarseks epidemioloogiaks. Juhul kui analüüsi kaasatakse patogeenide genoomid juba täispikkuses, siis võib seda nimetada genoomide epidemioloogiaks. Genoomide epidemioloogia kohta on lähiminevikust võtta mitmeid häid näiteid.
Aastail 2013–2016 Lääne-Aafrikas möllanud Ebola viiruse puhangu korral demonstreeriti tänu täispikkuses sekveneeritud genoomide analüüsile, et kogu puhang on põhjustatud ühest loomadelt üle kandunud tüvest. Veidi hiljem aga seda, et Ebola viirus võib varjatult püsida varem haigust põdenud inimeses ning seejärel seksuaalselt levida ja aluse panna uuele puhangule. Nimelt leiti kaks sarnast Ebola viiruse genoomi, esimene varem Ebolast tervenenud mehe spermast ning teine mehega seksuaalsuhtes olnud ja pea pool aastat hiljem Ebolasse nakatunud naise verest.
Samuti suudeti Zika-viiruse puhul tänu täispikkuses genoomide analüüsile näidata, millised oli Lõuna-Ameerikas selle viiruse levikuteed ning seda, et epideemia sai alguse Põhja-Brasiiliast.
Eestiski on varem genoomide epidemioloogiaga tegeletud ja seda peamiselt Eesti HIV-1 epideemia analüüsil. Eestis on viiruste (HIV, HCV) sekveneerimise ja selle käigus kogutud andmete analüüsiga tegelenud HIV-töögrupp Tartu Ülikooli bio- ja siirdemeditsiini instituudi mikrobioloogia osakonnas. Töögrupp on 15 aastat tegelenud peamiselt HIV uuringutega, millest suur osa hõlmab viiruse sekveneerimist ja selle tulemusel viiruse mitmekesisuse ja HIV ravimresistentsuse määramise ja analüüsimisega. Kokku viis üle-eestilist uuringut on keskendunud HIV ülekanduva ravimresistentuse (inimesed on nakatunud HIV ravimitele resistentsete viirustega) määramisele ja jälgimisele ajas. Need uuringud on näidanud, et Eestis näitab HIV ülekanduv ravimresistentsus ajas tõusvat trendi, kuid püsib suhteliselt madalal (alla 8%). Lisaks sellele on uuringud kirjeldanud Eesti HIV molekulaarepidemioloogiat aastate jooksul. Nendest uuringutest selgus, et erinevalt naaberriikidest levivad Eestis tõenäoliselt Lääne-Aafrikast pärit HIV-1 CRF06_cpx viirused, mis vähemal määral on omakorda segunenud nn Ida-Euroopas levinud HIV-1 alatüübi A6 viirustega.
SARS-CoV-2 SEKVENEERIMINE EESTIS
Eesti COVID-19 epideemia kontekstis toimuv SARS-CoV-2 genoomide sekveerimine ja analüüs on juurutatud algselt Tartu Ülikooli (TÜ) tehnoloogiainstituudis ning hetkel leiab aset peamiselt KoroGeno-EST projektide raames TÜ bio-ja siirdemeditsiini instituudi mikrobioloogia osakonnas (CTM 2021). Ülalmainitud varasemad eksperditeadmised HIV molekulaar-epidemioloogia alal lõid suurepärase võimaluse kiireks fokusseerumiseks SARS-CoV-2 genoomide sekveerimisele ja analüüsile. Protsess ise saab alguse viirusnakkuse suhtes positiivseks diagnoositud proovidest, millest koostatakse valim koostöös Terviseametiga. Proovis olevast bioloogilisest materjalist eraldatakse viiruse RNA (Synlab Eesti), see konverteeritakse DNAks, paljundatakse ja järjestatakse süvasekveneerimismeetodil TÜ genoomika instituudi tuumiklaboris. Seejärel liiguvad sekveneerimisel saadud andmed TÜ teadusarvutuste keskuse arvutusklastrisse, kus neile tehakse bioinformaatiline analüüs. Bioinformaatilise analüüsi käigus leitakse mutatsioonid, mis eristavad proovis olevat viirust Hiinas epideemiale alguse pannud viirusest. 14 kuud pärast epideemia algust on keskmise viiruse erinevus algsest Wuhani tüvest keskmiselt 30-40 nukleotiidi ehk mutatsiooni. Leitud mutatsioonide mustri põhjal on võimalik määrata, millisesse varianti või tüvesse viirus kuulub ning milline on ta sugulus (geneetiline distants) teiste SARS-CoV-2 viirustega.
MAAILMA JA EESTI SARS-CoV-2 MOLEKULAARNE EPIDEMIOLOOGIA
SARS-CoV-2 sekveneerimisprojektide fookus on epideemia jooksul veidi muutunud. Epideemia alguses oli põhirõhk koostöös Terviseametiga nakkuskollete geneetilisel analüüsil. Analüüsi kasutati peamiselt tundmatut päritolu nakkuse saanute seostamiseks kindlate nakkuskolletega ning nakkuskollete omavaheliste seoste kindlakstegemiseks. Selleks kasutati evolutsioonibioloogias laialt kasutatavat fülogeneetilist analüüsi. Näitena võib tuua analüüsi, kuidas olid seotud Ukrainast Tallinnasse saabunud bussireisijad Nõo alevikus aset leidnud puhanguga. Mitmetel juhtudel tehti kindlaks ka see, kas (ravi)asutustes aset leidnud puhangud on sinna sisse toodud mitu korda eraldi või oli tegu majasisese levikuga. Viimane teadmine on hädavajalik viirusevastaste meetodite valikul. Üldiselt on fülogeneetiline analüüs näidanud, et nakkuskoldeid kipuvad moodustama pigem viirused Eesti eri piirkondadest koos Tallinnaga, mitte aga Eesti eri piirkondade viirused omavahel.
2020. aasta lõpust alates on SARS-CoV-2 molekulaarepidemioloogia fookus veidi nihkunud nakatunute arvu tõusu ning erinevalt käituvate SARS-CoV-2 tüvede ning variantide ilmumise tõttu: projekti põhieesmärgiks sai erinevate SARS-CoV-2 genotüüpide määramine erinevates nakatunute riskigruppides. Kuigi riskigruppide definitsioonid ning nende suhtelised suurused sõltuvad paljuski riigi epidemioloogilisest olukorrast, võib siinkohal välja tuua üldjoontes järgnevad kolm jaotust: esimeseks on juhuvalim nakatunutest sh tundmatut päritolu nakatunud. See grupp annab ülevaate erinevate tüvede ja variantide üldisest levikust riigis; teiseks on välismaal nakatunud või nendega seotud nakatunud. See annab aimu, millised tüved ning millistest peamistest reisi sihtkohtadest Eestisse saabuvad. Teatud variantide prevaleerimise korral lubab see ka ennustada eelseisvate nakkuste iseloomu. Kolmandaks vaktsineeritud nakatunud. Andmed selles grupis levivatest viiruste mutatsioonidest ning nende genotüüpidest lubavad hinnata, millised on erisuguste vaktsiinide efektiivsused ning millise tõenäosusega võivad tekkida või levida tüved, kes suudavad vaktsiinide tekitatud immuunkaitsest läbi murda.
KoroGeno-EST projektide raames on kokku sekveneeritud üle 2800 viiruse täisgenoomi. Nende fülogeneetiline analüüs näitas, et epideemia alguses oli Eesti SARS-CoV-2 genotüüpide struktuur mitmekesine ning sarnanes üldjoontes sellele, mida nähti samal ajal teistes Euroopa riikides. Enim levinud olid B.1.5, B.1.1 ja B.1.1.10 genotüübid. Samuti oli sage mujal riikides haruldasem, kuid ka näiteks Tšehhis levinud variant B.1.258. See tüvi paistab silma Inglise tüvega identse S-geenis asuva deletsiooni S:del69/70 poolest. Veebruarist alates leidis Eestis nagu ka ülejäänud Euroopas aset SARS-CoV-2 variantide muutus. Veebruari keskpaigast kuni märtsi alguseni tõusis kiirelt Inglise tüve osakaal (B.1.1.7), mis tõrjus välja kõik teised tüved, sealhulgas B.1.258. Aprilli alguseks oli Inglise tüvi saavutanud pea 80–90protsendilise leviku. Kliiniliselt ja immunoloogiliselt olulise LAV tüve (B.1.351) üksikuid juhte hakati registreerima märtsis-aprillis ning mai alguse seisuga on leitud kokku alla 50 juhu, mis moodustas paar protsenti sekveneeritud juhtudest. Enamik neist on seotud välisriikides nakatumistega. Tänaseks on teada ka üks peamiselt Lõuna-Eestiga seotud LAV tüve kolle. SARS-CoV-2 eri genotüüpide osakaalu muutust Eestis 2021. aastal illustreerib joonis 1.
Joonis 1. Erinevate SARS-CoV-2 genotüüpide osakaalud Eestis jaanuari keskelt mai alguseni 2021. aastal. Kuni veebruari II nädalani oli valdav enamus UK tüvedest sissetoodud
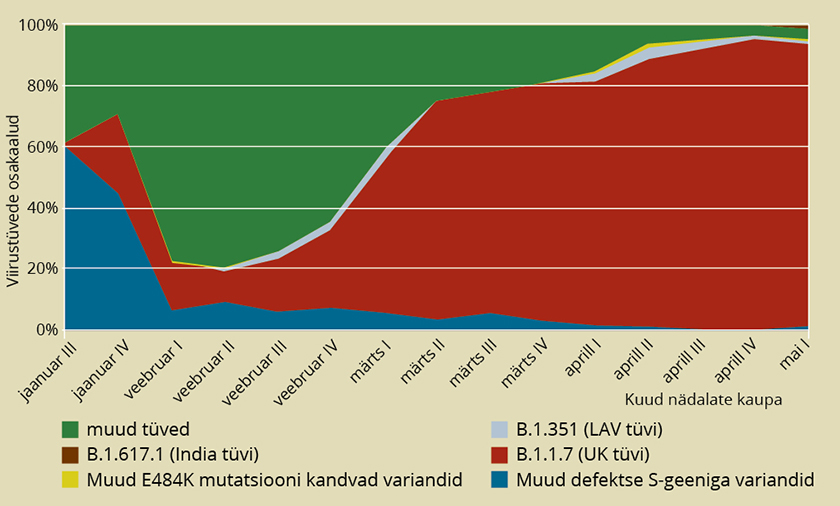
Allikas: Autorite koostatud
LÕPPSÕNA
Tõenäoliselt jääb SARS-CoV-2 täisgenoomide sekveneerimine veel pikemaks ajaks oluliseks meetodiks SARS-CoV-2 epideemia ohjamisel. Kindlasti kasvab SARS-CoV-2 täisgenoomide järjestuste tähtsus vastavalt selle, kuidas reageerib ülemaailmne epideemia mitmesuguste vaktsiinide kasutuselevõtule. Juhul kui mingitel vaktsiinidel peaks ilmnema oluline SARS-CoV-2 variantide spetsiifiline mõju või viirus suudab edaspidi jooksvalt tekitada vaktsiinide eest pagemise mutatsioone, saab SARS-CoV-2 täisgenoomide sekveneerimine ilmselt möödapääsmatuks osaks riikide vaktsineerimispoliitikas. Võimalik ka, et vaktsiinid muutuvad maailmas piirkondadepõhiseks, mis tekitab jällegi vajaduse antud piirkonnas läbi viia SARS-CoV-2 viiruste genotüpeerimine ja kaardistamine. Oma osa sellesse võib edaspidi lisada ka reoveest SARS-CoV-2 variantide osakaalu määramine.
Tõenäoliselt kasutatakse SARS-CoV-2 täisgenoomide järjestamist veel pikka aega olulise meetodina koroonaepideemia ohjamisel.
COVID-19 epideemia on esitanud molekulaar-epidemioloogiale seninägematu väljakutse. Samas on tänapäeval molekulaarbioloogia tööriistad võimsamad kui kunagi varem. Loodame, et molekulaar-epidemioloogid nii siin kui ka sealpool piiri suudavad vastata järjest enamatele COVID-19 epideemia püstitatud küsimustele. Samuti loodame, et kui kunagi peaks puhkema mõne uue tundmatu nakkuse epideemia, on meil praegusest olukorrast kaasa võtta piisavalt kogemusi.
Tänusõnad
Täname kõiki projekti meeskonna liikmeid, ilma kelleta poleks projekt teoks saanud:
- TÜ mikrobioloogia osakond: Irja Lutsar, Radko Avi, Kristi Huik, Taavi Päll, Merit Pauskar, Arina Shablinskaja, Ene-Ly Jõgeda, Eveli Kallas, Kai Truusalu, Dagmar Hoidmets
- TÜ tehnoloogia instituut: Aare Abroi
- TÜ genoomika instituut: Tuuli Reisberg, Mait Metspalu, Lili Milani
- TÜ teadusarvutuste keskus: Ulvi Talas
- Terviseamet: Mari-Anne Härma, Olga Sadikova, Jevgenia Epštein, Liidia Dotsenko, Heiki Niglas, Hanna Sepp, Mats Hansen, Tiia Luht, Kaisa Kuus
- TÜ Kliiniliste Uuringute Keskus: Katrin Kaarna, Evelin Lehtoja, Freddy Lättekivi, Kerli Meister
- Synlab Eesti: Paul Naaber, Andrio Lahesaare, Kaspar Ratnik
KASUTATUD ALLIKAD
- CTM (2021). Eesti SARS-CoV-2 täisgenoomide järjestamine ja analüüs. KoroGeno-EST-1/EST-2. Kokkuvõte geeniuuringust. Riiklik siirdemeditsiini ja kliiniliste teadusuuringute keskus. – ctm.ee